Clinical trials are research studies that aim to determine whether a medical strategy, treatment, or device is safe for use or consumption by humans.
These studies may also assess how effective a medical approach is for specific conditions or groups of people.
Overall, they add to medical knowledge and
To ensure participant safety, trials start with small groups and examine whether a new method causes any harm or unsatisfactory side effects. This is because a technique that is successful in a laboratory or in animals may not be safe or effective for humans.
Fast facts on clinical trials
- Clinical trials aim to find out whether a medical strategy, treatment, or device is safe and effective for humans to use or consume.
- Trials consist of four phases, and they can focus on: treatment, prevention, diagnostic, screening, supportive care, health services research, and basic science.
- A research team will likely include doctors, nurses, social workers, health care professionals, scientists, data managers, and clinical trial coordinators.
- Participation can involve both risks and benefits. Participants must read and sign the “informed consent” document before joining a trial.
- Risks are controlled and monitored, but the nature of medical research studies means that some risks are unavoidable.
The main purpose of clinical trials is research. Trials are designed to add to medical knowledge related to the treatment, diagnosis, and prevention of diseases or conditions.

Studies follow strict scientific standards and guidelines that aim to:
- protect participants
- provide reliable and accurate results
Clinical trials on humans occur in the final stages of a long, systematic, and thorough research process.
The process
Testing on animals enables scientists to see how the approach affects a living body.
Finally, human testing is carried out in small and then larger groups.
Trials may be carried out to:
- Evaluate one or more treatment interventions for a disease, syndrome or condition, such as drugs, medical devices, or approaches to surgery or therapies
- Assess ways to prevent a disease or condition, for example, through medicines, vaccines, and lifestyle changes
- Evaluate one or more diagnosis interventions that might identify or diagnose a particular disease or condition
- Examine identification methods for recognizing a condition or risk factors for that condition
- Explore supportive care procedures to improve the comfort and quality of life of people with a chronic illness
The outcome of a clinical trial may identify if a new medical strategy, treatment or device:
- has a positive effect on patient prognosis
- causes unforeseen harm
- has no positive benefits or has negative effects
Clinical trials can provide valuable information regarding the cost-effectiveness of a treatment, the clinical value of a diagnostic test, and how a treatment improves quality of life.
All clinical trials have a primary purpose. These can be broken down into the following categories:
- Treatment: Testing new treatments, new drug combinations, or new approaches to surgery or therapy
- Prevention: Examining ways to improve prevention or recurrence of disease through, for example, medicines, vitamins, vaccines, minerals, and lifestyle changes
- Diagnostic: Finding improved testing techniques and procedures for diagnosing diseases and conditions
- Screening: Testing the best method of identifying certain diseases or health conditions
- Supportive care: Investigating procedures to improve comfort and quality of life for patients with a chronic condition
- Health services research: Evaluating the delivery, process, management, organization, or financing of health care
- Basic science: Examining how an intervention works
Clinical trials help improve and advance medical care. The studies provide factual evidence that can be used to improve patient care.
Clinical research is only conducted if doctors are unaware of elements such as:
- whether a new approach works effectively in humans and is safe
- what treatments or strategies work most successfully for certain illness and groups of individuals
Various elements are involved in setting up, running, and following up a clinical trial.
Clinical trials protocol
A trial follows a comprehensive plan, or protocol. A protocol is the written description of a clinical trial.
It includes the study’s objectives, design and methods, relevant scientific background, and statistical information.
Key information to include may be:
- the number of participants
- who is eligible to take part
- what tests will be given and how often
- types of data to be collected
- the length of the study
- detailed information about the treatment plan
Avoiding bias
Researchers must take measures to avoid bias.
Bias refers to human choices or other factors that are not related to the protocol but which may affect the results of the trial.
Steps that can help to avoid bias are comparison groups, randomization, and masking.
Comparison groups
Most clinical trials use comparison groups to compare medical strategies and treatments. Results will show if one group has a better outcome than the other.
This is usually conducted in one of two ways:
- One group receives an existing treatment for a condition, and the second group receives a new treatment. Researchers then compare which group has better results.
- One group receives a new treatment, and the second group receives a placebo, an inactive product that looks like the test product.
Randomization
Clinical trials with comparison groups often use randomization. Participants are allocated to comparison groups by chance rather than by choice. This means that any differences seen during a trial will be due to the strategy used and not because of pre-existing differences between participants.
Masking or blinding
Masking or blinding helps avoid bias by not informing either the participants or the researchers which treatment the participants will be receiving.
Single blind: This is when either the participants or researchers are unaware, of which group is which.
Double blind: This is when both participants and researchers are unaware.
Confounding factors
A confounder can distort the true relationship between two or more characteristics.
For example, one could conclude that people who carry a cigarette lighter are more likely to develop lung cancer because carrying a lighter causes lung cancer. Smoking is a confounder in this example.
People who carry a cigarette lighter are more likely to be smokers, and smokers are more likely to develop lung cancer, but some people may carry a lighter for other purposes.
Not taking this into consideration can lead to false conclusions.
Who is in the research team?
A principle investigator, who is usually a medical doctor, will lead each clinical study.
The research team may include:
- doctors
- nurses
- social workers
- health care professionals
- scientists
- data managers
- clinical trial coordinators
Where are clinical trials conducted?
The location will depend on the type of study and who is organizing it.
Some common locations include:
- hospitals
- universities
- medical centers
- doctors’ offices
- community clinics
- federally-funded and industry-funded research sites
How long do trials last?
This depends on what is being studied, among other factors. Some trials last days, while others continue for years.
Before enrolling in a trial, participants will be told how long it is expected to last.
There are different types of study, and different ways of organizing them. Here are some study types.
Observational studies
Cohort studies and case control studies are examples of observational studies.
Cohort study
A cohort study is an observational study in which the study population, or cohort, is selected.
Information is gathered to establish which subjects have either:
- a particular characteristic, such as a blood group that is thought to be related to the development of the disease in question
- exposure to a factor that may be linked to a disease, for example, cigarette smoking
An individual could be chosen because they smoke. They may then be followed forward in time to see how likely they are to develop a disease, compared with other people.
This type of study is used to study the effect of suspected risk factors that cannot be controlled experimentally, such as the impact of smoking on lung cancer.
The main advantages of cohort studies are:
- Exposure is measured in advance of disease onset and is therefore likely to be unbiased in terms of disease development.
- Rare exposures can be investigated by suitable selection of study cohorts.
- Multiple outcomes — or diseases — can be studied for any one exposure.
- Disease incidence can be calculated in both the exposed and unexposed groups.
The main disadvantages of cohort studies are:
- They tend to be expensive and time-consuming, especially if they are conducted prospectively, which means moving forward.
- Changes in both exposure status and diagnostic criteria over time can affect the classification of individuals according to exposure and disease status.
- There could be information bias in the concluded outcome because the subject’s exposure status is known.
- Losses to follow-up may present selection bias.
Case control studies
A case-control study can distinguish risk factors for a particular medical condition.
Researchers compare people with a condition and those without it. Working backward through time, they identify how the two groups differ.
Case-control studies are always retrospective — looking backward — because they begin with the outcome and then trace back to investigate exposures.
The
- Findings can be obtained quickly.
- The study can take place with a minimum of funding or sponsorship.
- They are efficient for investigating rare diseases or diseases with a long induction period.
- A wide range of possible risk factors can be examined.
- Multiple exposures can be studied.
- They require few study subjects.
The main disadvantages of case-controlled studies are:
- Incidence data cannot be generated.
- They are subject to bias.
- It can be difficult to obtain accurate, unbiased measures of past exposures if record keeping is inadequate or unreliable. This is called information bias.
- Selection of controls can be problematic. This may introduce selection bias.
- The chronological sequence between exposure and disease may be hard to identify.
- They are not appropriate for examining rare exposures, unless the exposure is responsible for a large percentage of cases.
Nested case-control study
In a nested case-control study, the groups — cases and controls — come from the same study population, or cohort.
As the cohort is followed forward, the cases that arise become the “cases” in the case-control study. The unaffected participants of the cohort become the “controls.”
Nested case-control studies are less costly and less time-consuming when compared with a cohort study.
Incidence and prevalence rates of the disease can occasionally be projected from a nested case-control cohort study. This is not possible from a simple case-control study, as the total number of exposed individuals and the follow-up times are usually unknown.
The main advantages of nested case-control studies are:
- Efficiency: Not all of the participants of the cohort require diagnostic testing.
- Flexibility: They allow the testing of hypotheses that were not anticipated when the cohort was planned.
- Reduction of selection bias: Cases and controls are sampled from the same population.
- Reduction of information bias: Risk factor exposure can be assessed with the investigator blind to case status.
The main disadvantage is that the results have lower authority, due to the small sample size.
Ecological study
An ecological study looks at the relationship between exposure and outcome of the population or community.
Common categories of ecological study include:
- geographical comparisons
- time-trend analysis
- studies of migration
The main advantages of ecological studies are:
- They are inexpensive, as routinely collected health data can be utilized.
- They are less time-consuming than other studies.
- They are uncomplicated and straightforward to understand.
- The effect of exposures that are measured over groups or areas — such as diet, air pollution, and temperature — can be investigated.
The main disadvantages of ecological studies are:
- Errors of deduction known as ecological fallacy can occur. It happens when researchers draw conclusions about individuals based solely on the analysis of group data.
- Exposure to outcome relationships is difficult to detect.
- There is a lack of information on confounding factors.
- There may be systematic differences between areas in how exposures are measured.
Experimental studies
Apart from observational studies, there are also experimental studies, including treatment studies.
Randomized controlled trials
A randomized controlled trial (RCT) randomly allocates individuals either to receive or not receive a particular intervention.
One of two different treatments will be used, or a treatment and a placebo.
This is the most effective study type for identifying which treatment works best. It reduces the influence of external variables.
The main advantages of RCTs are:
- There is no conscious or subconscious bias on the part of the researcher. This essentially guarantees external validity.
- Confounding variables such as age, gender, weight, activity level, and so on, can be canceled out, as long as the sample group is large enough.
The main disadvantages of RCTs are:
- They are time-consuming.
- They can be expensive.
- They require large sample groups.
- Rare events can be difficult to study.
- Both false-positive and false-negative statistical errors are possible.
Adaptive clinical trial
An adaptive design method is based on collected data. It is both flexible and efficient. Modifications can be made to the trial and the statistical procedures of ongoing clinical trials.
Quasi-experiment
Quasi-experimental, or “nonrandomized” studies, include a broad range of intervention studies that are not randomized. This type of
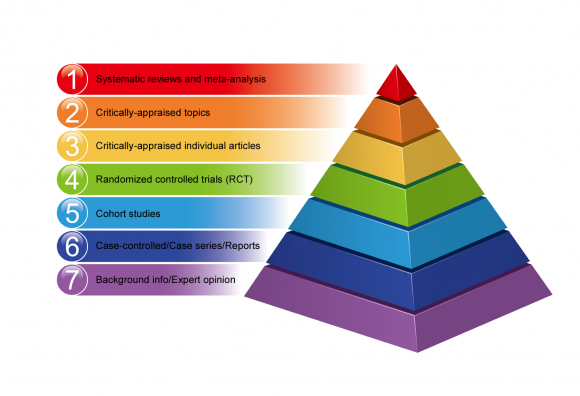
A number of hierarchies of evidence have been founded to enable various research methods to be ranked according to the validity of their findings.
Hierarchies of evidence make it possible to rank various research methods according to the validity of their findings.
Not all research designs are equal in terms of the risk of error and bias in their results. Some methods of research provide better evidence than others.
Below is an example of the hierarchy of evidence-based medicine
Medical research studies are divided into different stages, called phases. For drug testing, these are defined by the FDA.
Early phase trials
Phase 0 trials: Pharmacodynamics and pharmacokinetics
Phase 0 is an exploratory phase that helps provide clinical information for a new drug at an earlier phase.
- is conducted early in phase 1
- involves very limited human exposure
- has no therapeutic or diagnostic intent, being limited to screening and microdose studies
Phase 1 trials: Screening for safety
After phase 0, there are four more phases of trials in humans. These often overlap. Phases 1 through 3 take place before a license is granted.
Phase 1 guidelines involve:
- between 20 and 80 healthy volunteers
- verification of the most frequent side effects of the drug
- finding out how the drug is metabolized and excreted
Phase 2 trials: Establishing effectiveness
If phase 1 studies do not reveal unacceptable toxicity levels, phase 2 studies can begin.
This involves:
- between 36 and 300 participants
- collecting preliminary data on whether the drug works in people with a certain disease or condition
- controlled trials to compare those receiving the drug with people in a similar situation who are receiving a different drug or a placebo
- continued safety evaluation
- studies of short-term side effects
Phase 3 trials: Final confirmation of safety and effectiveness
If phase 2 has confirmed the effectiveness of a drug, the FDA and sponsors will discuss how to conduct large-scale studies in phase 3.
This will involve:
- between 300 and 3,000 participants
- gathering further information on safety and effectiveness
- studies of different populations
- examining various dosages to determine the best prescription amount
- using the drug in combination with other drugs to determine effectiveness
After this phase, the complete information on the new drug is submitted to the health authorities.
Review meeting
If the FDA approve the product for marketing, post-marketing requirement and commitment studies are conducted.
The FDA
New Drug Application
A drug sponsor will complete a New Drug Application (NDA) to ask the FDA to consider approving a new drug for marketing in the U.S.
An NDA
- all animal and human data
- analysis of data
- information regarding drug behavior in the body
- manufacture details
The FDA has 60 days to decide whether to file it to be reviewed.
If they decide to file the NDA, the FDA review team is assigned to evaluate the sponsor’s research on the drug’s safety and effectiveness.
The following steps must then take place.
Drug labeling: The FDA reviews the drug’s professional labeling and confirms appropriate information is shared with consumers and health professionals.
Facility inspection: The FDA inspect the facilities where the drug will be manufactured.
Drug approval: FDA reviewers either approve the application or issue a response letter.
Phase 4 trials: Studies during sales
Phase 4 trials take place after the drug has been approved for marketing. They are designed to include:
- over 1,000 patients
- comprehensive experience in evaluating the safety and effectiveness of the new medicine in a larger group and subpopulations of patients
- comparison and combination with other available treatments
- evaluation of long-term side effects of the drug
- detection of less common adverse events
- cost-effectiveness of drug therapy compared with other traditional and new therapies
Safety report
After the FDA approves a drug, the post-marketing stage begins. The sponsor, usually the manufacturer, submits periodic safety updates to the FDA.
Clinical trials and research can cost hundreds of millions of dollars. Groups that fund trials may include:
- pharmaceutical, biotechnology, and medical device companies
- academic medical centers
- voluntary groups and foundations
- National Institutes of Health
- government departments
- physicians and health providers
- individuals
The protocol defines who is eligible to participate in a trial.
Possible inclusion criteria may be:
- having a specific illness or condition
- being “healthy,” with no health condition
Exclusion criteria are the factors that exclude some people from joining a trial.
Examples include age, gender, a specific type or stage of a disease, previous treatment history, and other medical conditions.
Taking part in clinical trials can have
Possible benefits of clinical trials include the following:
- Participants have access to new treatments.
- If a treatment proves successful, participants will be among the first to benefit.
- Participants who are not in the group receiving a new treatment may receive the standard treatment for the particular condition, which may be as good or better than the new approach.
- Health is closely monitored and supported by a team of health providers.
- Information gathered from clinical trials adds to scientific knowledge, may help others, and ultimately improves health care.
Possible risks include:
- Standard care for a particular condition can sometimes be better than the new strategy or treatments being studied.
- The new approach or treatment may work well for some participants but not necessarily for others.
- There may be unexpected or unforeseen side effects, especially in phase 1 and phase 2 trials and with approaches such as gene therapy or new biological treatments.
- Health insurance and health providers do not always cover patient care and costs for those participating in clinical trials.
The informed consent document explains the risks and potential benefits of taking part in a clinical trial.
Elements that must appear in the document include, among others:
- purpose of research
- foreseeable risks of discomforts
- possible benefits
Participants are expected to
The FDA works to ensure that anyone who is considering joining a trial has access to all the reliable information they need to make an informed choice, including information about the risks.
While risks to participants are controlled and monitored, some risks may be unavoidable, due to the nature of medical research studies.
Safety of participants is a high priority issue. In every trial, scientific oversight and patient rights contribute to their protection.
Good clinical practice (GCP)
GCP compliance provides the public with confidence that the safety and rights of participants are protected.
It aims to:
- to protect the rights, safety, and welfare of participants
- to guarantee that data collected is reliable, has integrity, and is of an appropriate quality
- to provide guidelines and standards for the conduct of clinical research
The foundations of GCP were first laid out in 1947. The main points were that, during any trials, researchers must guarantee:
- voluntary participation
- informed consent
- minimization of risk
Over time, additions have ranged from establishing additional protection for vulnerable populations to providing guidance to bodies carrying out research.
Patient rights
Ways of protecting patient rights include the following:
Informed consent is the process of supplying clinical trial participants with all of the facts about the trial. It happens before the participants agree to take part and during the course of the trial. Informed consent includes details about the treatments and tests that may be received and the possible benefits and risks.
Other rights: The informed consent document is not a contract; participants may withdraw from the study at any time regardless of whether or not the trial is complete.
Rights and protection for children: A parent or legal guardian must give legal consent if the child is aged 18 years or younger. If a trial may involve a risk that is greater than minimal, both parents must give permission. Children over the age of 7 years must agree to be involved in clinical trials.
Information about current clinical trials can be found here.